基于NMRPipe的RDC数据处理教程
0. 预备知识
残余偶极耦合(Residual Dipolar Coupling,RDC)是一种基于核磁共振的表征手段,常用于蛋白质动态结构的辅助解析与验证,其原理为:空间上相互邻近,但不具备化学键连的偶极(如1H-15N原子对)之间,存在直接的电磁相互作用,在溶液中,偶极朝不同方向的分布相同,因而偶极-偶极相互作用平均值为零,无法直接观测;而在液晶或聚合物孔洞等定向程度高的环境,偶极朝不同方向的分布有差异,因此偶极-偶极相互作用平均值不为零(这是“残余(剩余)”一词的由来),表现为J耦合形成的裂分进一步增大或减小,其大小与偶极间的相对朝向有关,故可用于获取化学键的空间取向,以修正蛋白质结构。
通常情况下,测定RDC的常用核磁方法为IPAP-HSQC,这是一种自旋态选择性HSQC实验,通过在特定时间点交替施加和撤去脉冲,在消除直接维裂分的同时,将间接维裂分转变为方向相同的In-Phase分量和方向相反的Anti-Phase分量,再通过谱图加减,得到记录在不同图谱的高场分量与低场分量,避免信号峰相互叠加,从而大大降低RDC效应的测量难度,其原理大致如图1所示。
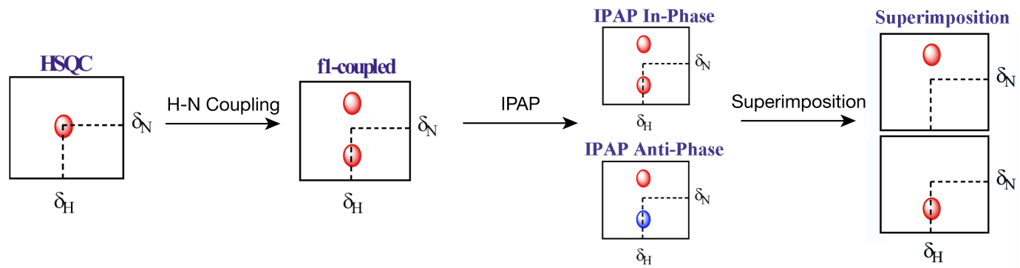
NMRPipe是由John Marino课题组与Ad Bax课题组联合开发的,适用于Unix及衍生系统的老牌核磁共振谱图处理软件。利用NMRPipe,可以把获得的原始IPAP谱图,拆分为独立的高场与低场分量,并能对拆分后的谱图进行相位调整、峰标记与峰归属(在预先准备归属文件的前提下)。
不过,由于NMRPipe并未将所有功能集成在一个GUI界面,而是采用命令行与简陋GUI相结合的方式,这对习惯于Windows的用户而言,无疑增加了操作难度,因此,笔者试着整理了一份基于NMRPipe的RDC数据处理教程,希望对NMRPipe新人有所帮助。
I. NMRPipe的安装
1. 安装包下载:访问NMRPipe官网,下载四个文件:NMRPipeX.tZ(NMRPipe源文件)、s.tZ(NMRPipe补丁和更新文件)、install.com(安装脚本)、binval.com(配合安装脚本使用的脚本)。如有需要,可以下载另外几个补充包:dyn.tZ(似乎用于在TALOS+中绘制拉氏图,同时存有DYNAMO and MFR),talos_nmrPipe.tZ(TALOS-N, TALOS+, and SPARTA+的必要运行文件),plugin.smile.tZ(SMILE NUS谱图重构程序)。
2. 将终端切换为C-shell:输入chsh -s /bin/tcsh,回车(没有tcsh/csh的情况下,首先用sudo yum(for RHEL-based Linux)/apt-get(for Ubuntu) install -y)。
3. 安装运行时必要的库文件:若为Cent OS 7,首先以root权限登录,找到/etc/yum.conf,添加multilib_policy=all并保存,然后切换为普通用户,分别输入以下命令并回车:
sudo yum install -y xterm
sudo yum install -y libgcc
sudo yum install -y glibc
sudo yum install -y libX11.so.6
sudo yum install -y libXext
sudo yum install -y libstdc++
sudo yum install -y xorg-x11-fonts-75dpi
sudo yum install -y xorg-x11-fonts-ISO8859-1-75dpi
这样就安装好运行时必要的库文件
(补充于2022.10)若为Ubuntu,所需的软件包和对应的名称与Cent OS有显著不同,因此运行的命令改为
sudo apt-get install tcsh
sudo apt-get install xterm
sudo apt-get install lib32z1
sudo apt-get install libx11-6:i386
sudo apt-get install libxext6:i386
sudo apt-get install xfonts-75dpi
sudo apt-get install msttcorefonts
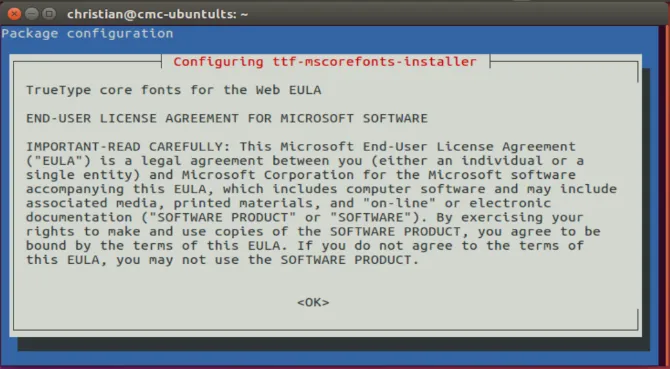
要注意的是,安装msttcorefonts时,在命令行下会弹出一个EULA协议的窗口,类似于下图所示,此时需要按Tab键,让光标转到《OK》上,然后回车,接下来按方向键,使光标停在《YES》上,回车即可。
4. 更改文件读写权限并安装:将四个文件放入指定文件夹中(如/home/(你的用户名)/NMRPipe),在该文件夹打开终端,输入:
chmod a+r *.tZ *.Z *.tar
chmod a+rx *.com
然后在当前terminal里输入xterm,回车;在新的xterm的窗口中输入./install.com,回车
5. 测试:退出终端,再重新打开终端,输入以下命令并回车,以测试NMRPipe运行情况:
which nmrPipe (Check that programs can be found)
nmrPipe -help (Run program in help mode)
man nmrPipe (Check manual pages)
bruker (varian delta) (Run the graphical conversion interface)
nmrDraw (Run the graphical interface)
若以上命令能正常运行,即表明NMRPipe安装成功
注意:按以上步骤安装,通常会将.cshrc一并配置,如果没有配置,请将以下代码粘贴至.cshrc
#!/bin/csh
if (-e /home/clemmensen/workbench/NMRPipe/com/nmrInit.linux212_64.com) then
source /home/clemmensen/workbench/NMRPipe/com/nmrInit.linux212_64.com
endif
if (-e /home/clemmensen/workbench/NMRPipe/com/font.com) then
source /home/clemmensen/workbench/NMRPipe/com/font.com
endif
II. 用NMRPipe处理IPAP-HSQC谱,并获取RDC数据
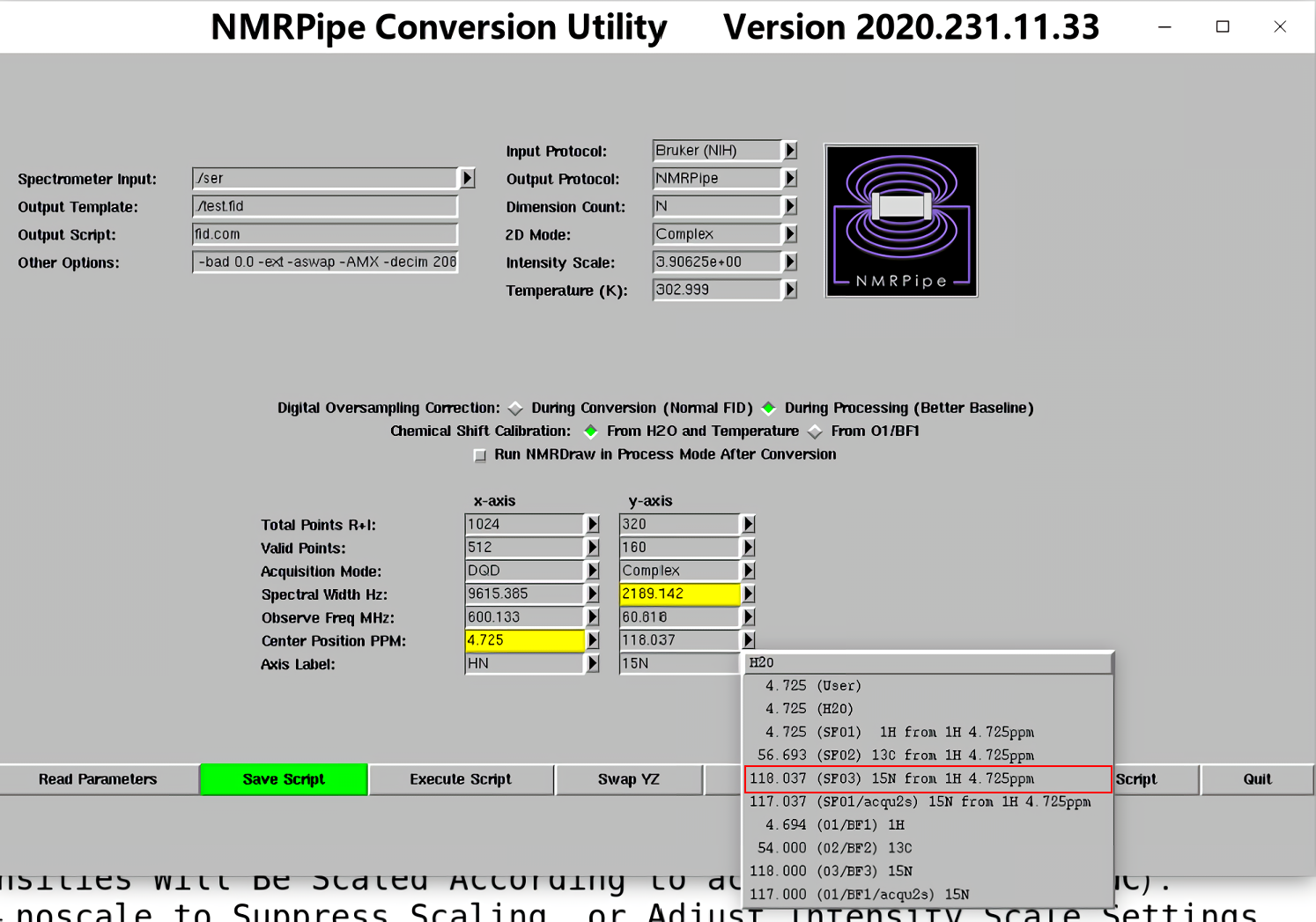
1.图谱格式转换:在原始图谱的文件夹中打开终端,输入bruker,回车,接下来会弹出如下窗口
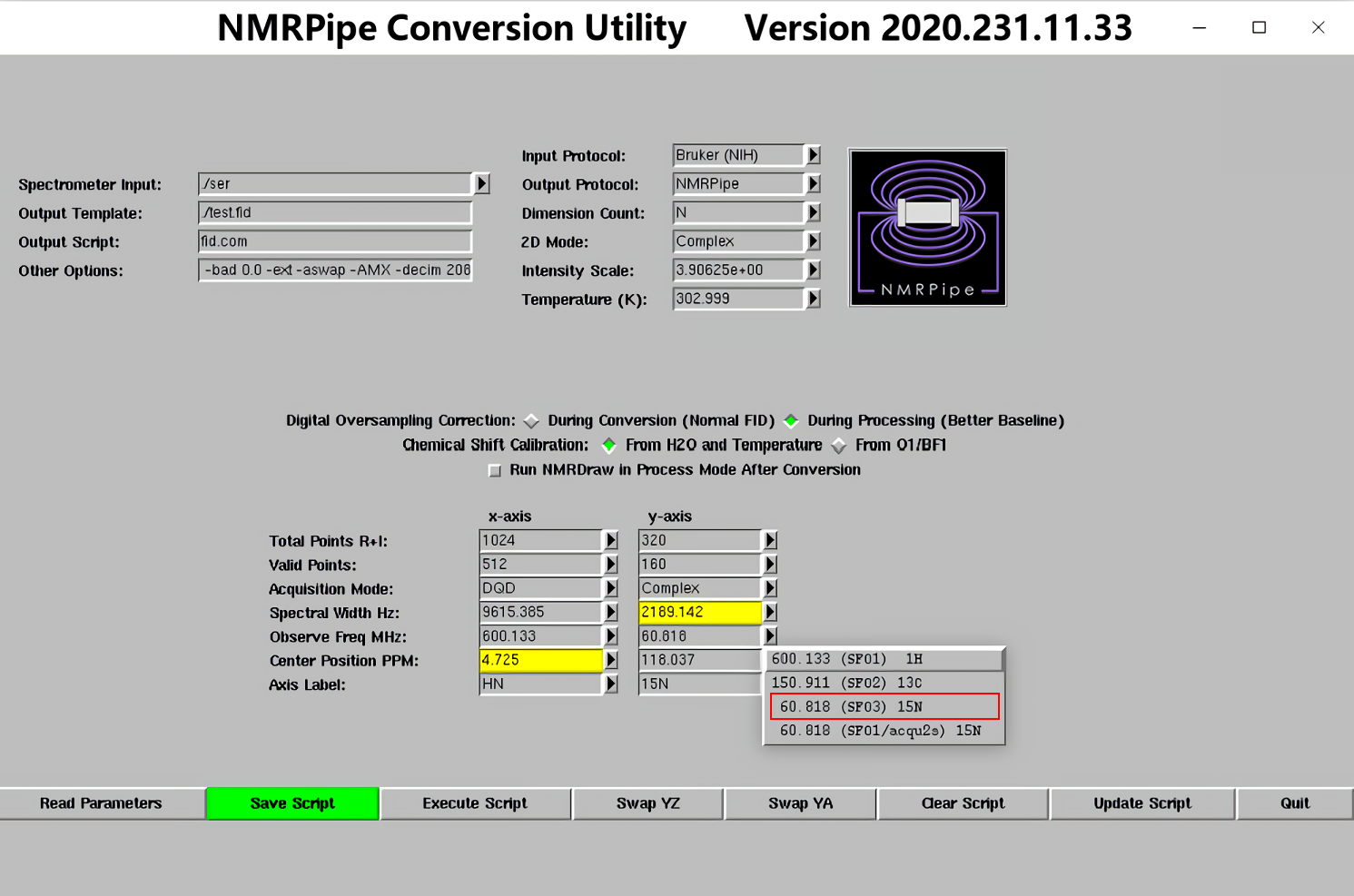
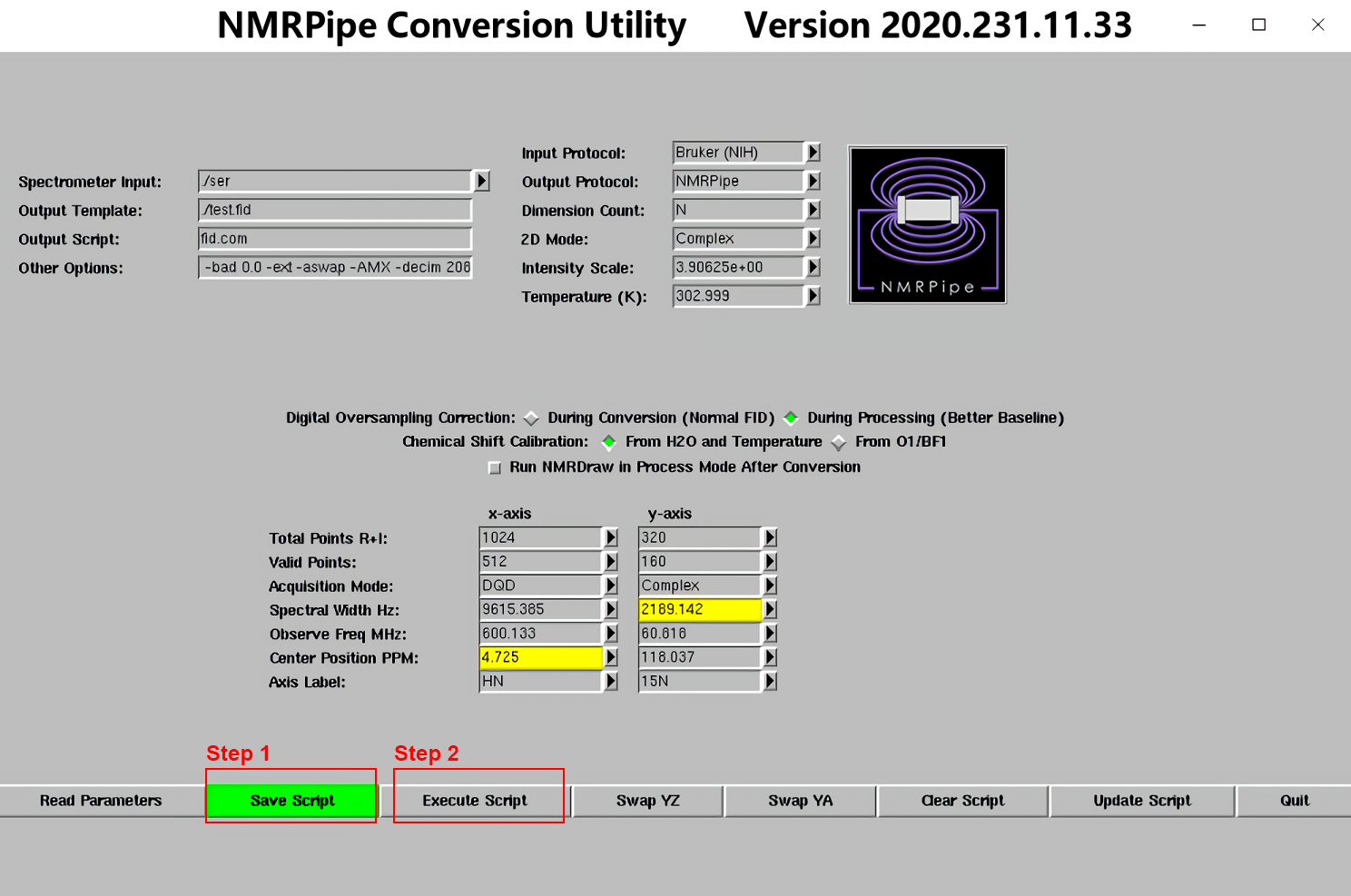
2.图谱的初步处理(HSQC及其衍生谱必做!)
将处理脚本hsqc.com复制到原始图谱的文件夹中,接着输入下列命令,赋予当前用户读写和运行脚本的权限,且能以可执行文件运行。(以后若不说明,所有.com脚本从本地上传至服务器后,或从Windows传至Linux虚拟机后,均执行此步骤)
chmod +755 hsqc.com
然后在终端中输入./hsqc.com,回车,得到初步处理后的二维图谱。
这样处理的图谱相位未必准确,所以我们要在NMRDraw中微调相位,使基线平齐,然后在hsqc.com中修改相位参数,再重复上述步骤。
在原始图谱的文件夹打开终端,输入nmrDraw,回车,会弹出如下窗口,此时右键单击“File”→“S) Select File”:
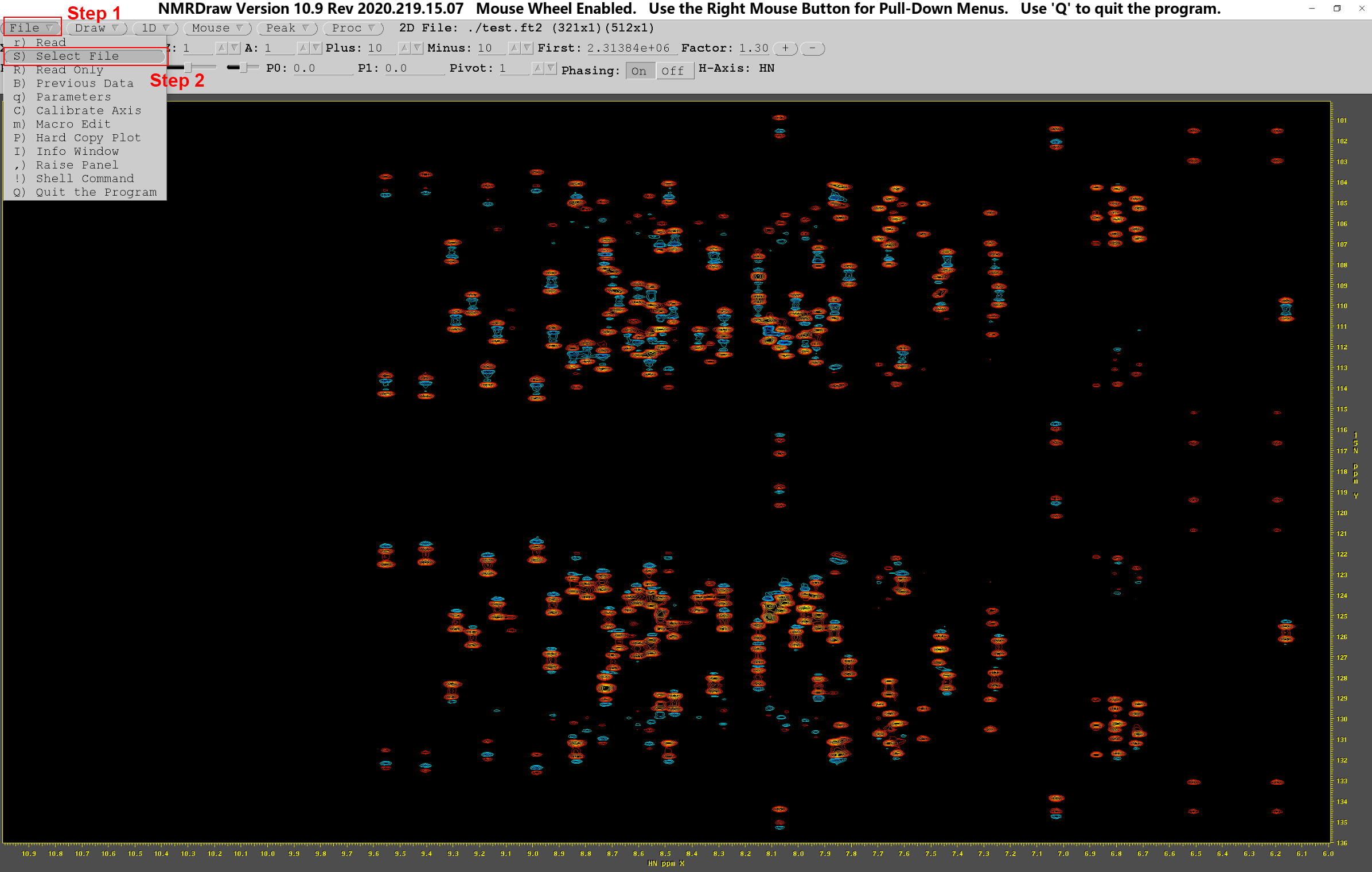
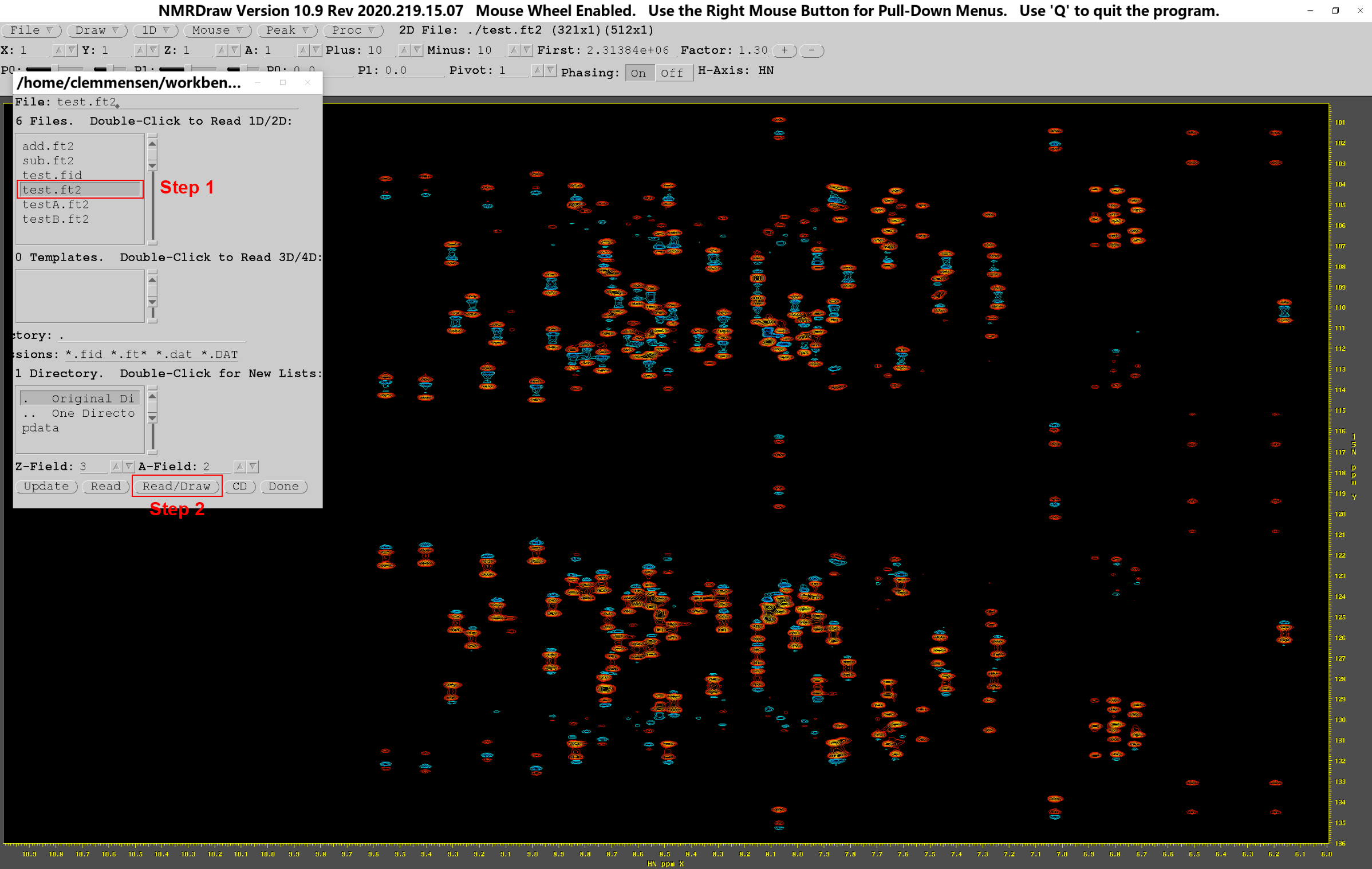
按H键,打开水平轴(1H)的NMR谱,点击图上信号明显的峰(尽量选取水平线上峰数较少的位置),便可查看点击位置水平线上的核磁信号;同理,按V键打开的是垂直轴(15N)的NMR谱。选取合适的基线参照位置后,可以拖动左上方工具栏的滑块(如红框所示),调节0阶和1阶校正的参数大小,或是直接输入具体数值调节。待基线平整,且信号峰显著,无杂峰干扰后,退出NMRDraw,修改.hsqc中相位校正的参数,然后重新运行.hsqc
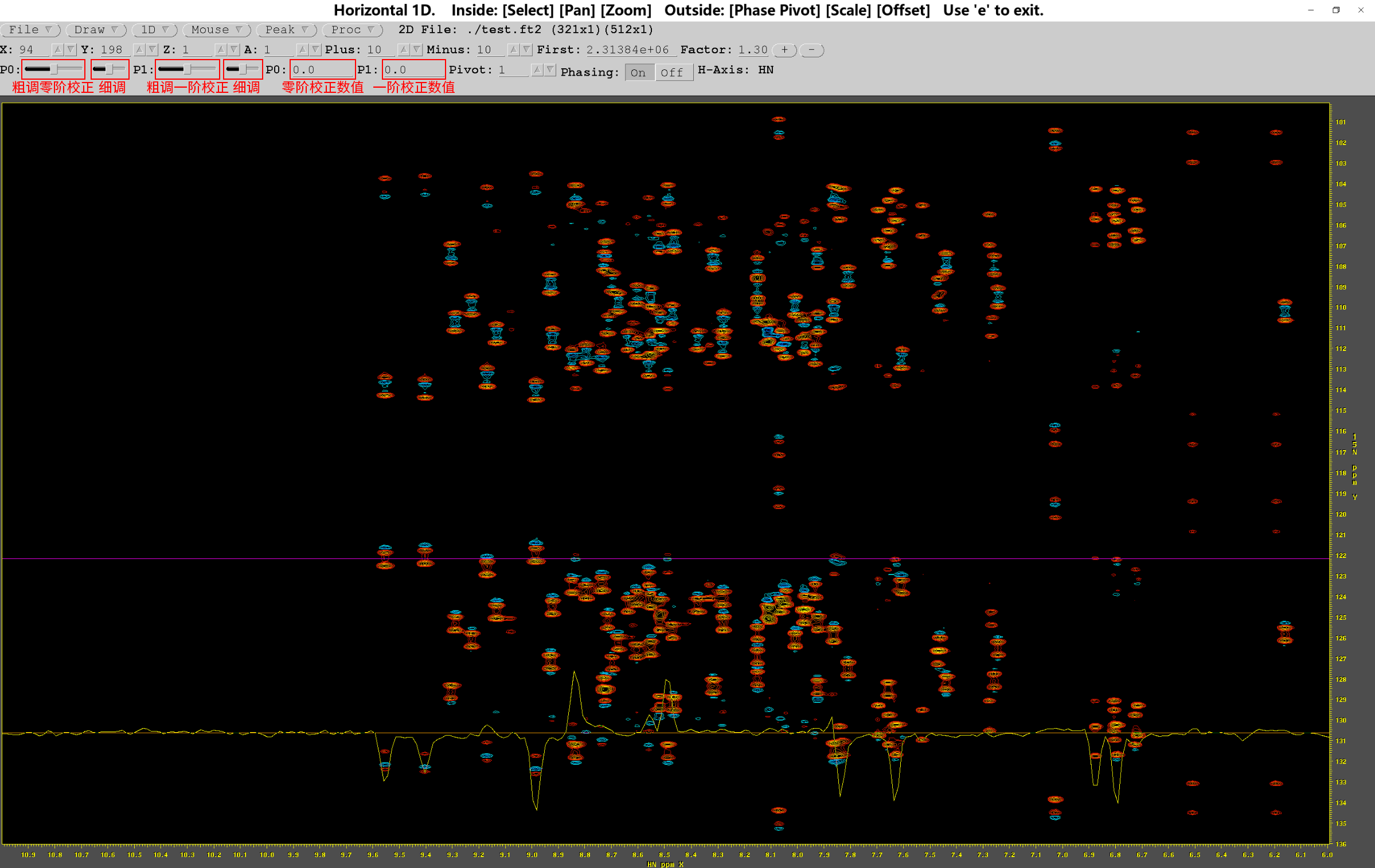
附:fid.com的代码(代码不唯一,以bruker命令生成的为准)
#!/bin/csh
bruk2pipe -verb -in ./ser \
-bad 0.0 -ext -aswap -AMX -decim 2080 -dspfvs 20 -grpdly 67.9842071533203 \
-xN 1024 -yN 320 \
-xT 512 -yT 160 \
-xMODE DQD -yMODE Complex \
-xSW 9615.385 -ySW 2189.142 \
-xOBS 600.133 -yOBS 60.818 \
-xCAR 4.725 -yCAR 118.037 \
-xLAB HN -yLAB 15N \
-ndim 2 -aq2D Complex \
| nmrPipe -fn MULT -c 3.90625e+00 \
-out ./test.fid -ov
sleep 5
附:hsqc.com的代码
#!/bin/csh
nmrPipe -in test.fid \ \
| nmrPipe -fn SP -off 0.5 -end 0.98 -pow 1 -c 1 \
| nmrPipe -fn ZF -auto \
| nmrPipe -fn FT \
| nmrPipe -fn PS -p0 -12.5 -p1 0 -di \
| nmrPipe -fn EXT -x1 11ppm -xn 6.0ppm -sw -verb \
| nmrPipe -fn TP \
| nmrPipe -fn SP -off 0.5 -end 0.98 -pow 1 -c 0.5 \
| nmrPipe -fn ZF -auto \
| nmrPipe -fn FT \
| nmrPipe -fn PS -p0 0 -p1 0 -di \
| nmrPipe -fn POLY -auto \
| nmrPipe -fn TP \
| nmrPipe -fn POLY -auto \
-verb -ov -out test.ft2
nmrPipe -in test.ft2| pipe2xyz -nv -out hsqc.nv -verb -ov
上述代码解释与注意事项:(1)-fn代表功能选项,SP代表窗函数,-pow为1时使用正弦窗函数,-pow为2时使用正弦方形窗函数,-c代表第一个点的放大倍数(?),ZF代表充零,FT代表傅里叶变换,PS代表相位校正,-p0为0阶校正,-p1为1阶校正,EXT代表提取部分图谱,-x1和-xn分别规定提取图谱的左右边界,TP代表数据转置(如x轴到y轴);傅里叶变换时默认从x轴(1H)开始处理
(2)第一处的相位校正调整的是1H的基线,第二处的相位校正调整的是15N的基线
(3)最后一行命令的目的,是将test.ft2通过pipe2xyz转换为NMRViewJ能识别的hsqc.nv,若不需要,可在前面加上#,这样该命令就会变成注释,不会自动执行
(4)有时候会发现初步处理的图谱如同从中间剪开后,再沿着没有剪的边拼合一般,此时我们应该把第二次出现的nmrPipe -fn FT改为nmrPipe -fn FT -alt
3.IPAP谱的分裂和谱图加减(IPAP谱必做!其他谱图跳过)
将处理脚本2p-pre.com复制到原始图谱的文件夹中,接着修改脚本权限,使脚本对所有用户均可读写执行,且能以可执行文件运行(以后涉及脚本处理时默认执行此步骤)。然后打开终端,输入./2p-pre.com,回车,生成分裂后的IPAP谱testA.ft2和testB.ft2。
接下来在终端输入如下代码并执行:
addNMR -in1 testA.ft2 -in2 testB.ft2 -add -out add.ft2
addNMR -in1 testA.ft2 -in2 testB.ft2 -sub -out sub.ft2
生成相加的图谱add.ft2和相减的图谱sub.ft2
同样要注意的是,按默认参数生成的add.ft2和sub.ft2相位可能存在较大偏差,导致最终在图上呈现的峰并非均一的单峰,甚至杂乱无章,此时需要按照第2部分所述调整水平轴(1H)和垂直轴(15N)的相位,然后在2p-pre.com中修改相位调整语句的参数,再重新运行2p-pre.com,最后对谱图加减。
附:2p-pre.com的代码
#!/bin/csh
nmrPipe -in test.fid \
| nmrPipe -fn COADD -time -axis Y -cList 1 0\
| nmrPipe -fn SP -off 0.5 -end 0.98 -pow 1 -c 1 \
| nmrPipe -fn ZF -auto \
| nmrPipe -fn FT \
| nmrPipe -fn PS -p0 180 -p1 0 -di \
| nmrPipe -fn EXT -x1 12ppm -xn 5.5ppm -sw -verb \
| nmrPipe -fn TP \
| nmrPipe -fn SP -off 0.5 -end 0.98 -pow 1 -c 0.5 \
| nmrPipe -fn ZF -auto \
| nmrPipe -fn FT -alt \
| nmrPipe -fn PS -p0 0 -p1 0 -di \
| nmrPipe -fn POLY -auto \
| nmrPipe -fn TP \
| nmrPipe -fn POLY -auto \
-verb -ov -out testA.ft2
nmrPipe -in test.fid \
| nmrPipe -fn COADD -time -axis Y -cList 0 1\
| nmrPipe -fn SP -off 0.5 -end 0.98 -pow 1 -c 1 \
| nmrPipe -fn ZF -auto \
| nmrPipe -fn FT \
| nmrPipe -fn PS -p0 180 -p1 0 -di \
| nmrPipe -fn EXT -x1 12ppm -xn 5.5ppm -sw -verb \
| nmrPipe -fn TP \
| nmrPipe -fn SP -off 0.5 -end 0.98 -pow 1 -c 0.5 \
| nmrPipe -fn ZF -auto \
| nmrPipe -fn FT -alt \
| nmrPipe -fn PS -p0 0 -p1 0 -di \
| nmrPipe -fn POLY -auto \
| nmrPipe -fn TP \
| nmrPipe -fn POLY -auto \
-verb -ov -out testB.ft2
#nmrPipe -in test.ft2| pipe2xyz -nv -out hsqc2.nv -verb -ov
部分代码解释:COADD等同于co-addition of data,暂时没搞清楚其参数的含义
4.对加减处理后的图谱标峰
以IPAP谱为例,我们需要给add.ft2和sub.ft2的信号峰标上序号,以便与标准谱图进行比较。
在当前终端输入nmrDraw,回车,按第2部分打开并读取add.ft2。如果怀疑当前图谱遗漏了重要的谱峰信号,可以点击“Factor”右边的“-”,减小信号显示阈值,然后点击“Draw”;如果感觉谱图杂峰较多,可以点击“Factor”右边的“+”,增大信号显示阈值,然后点击“Draw”;如果需要进一步微调,还可以直接在First右边的空格中,输入自己认为合适的检测阈值,回车后点击“Draw”。接下来,右键单击“Peak”→“K) Peak Detection”
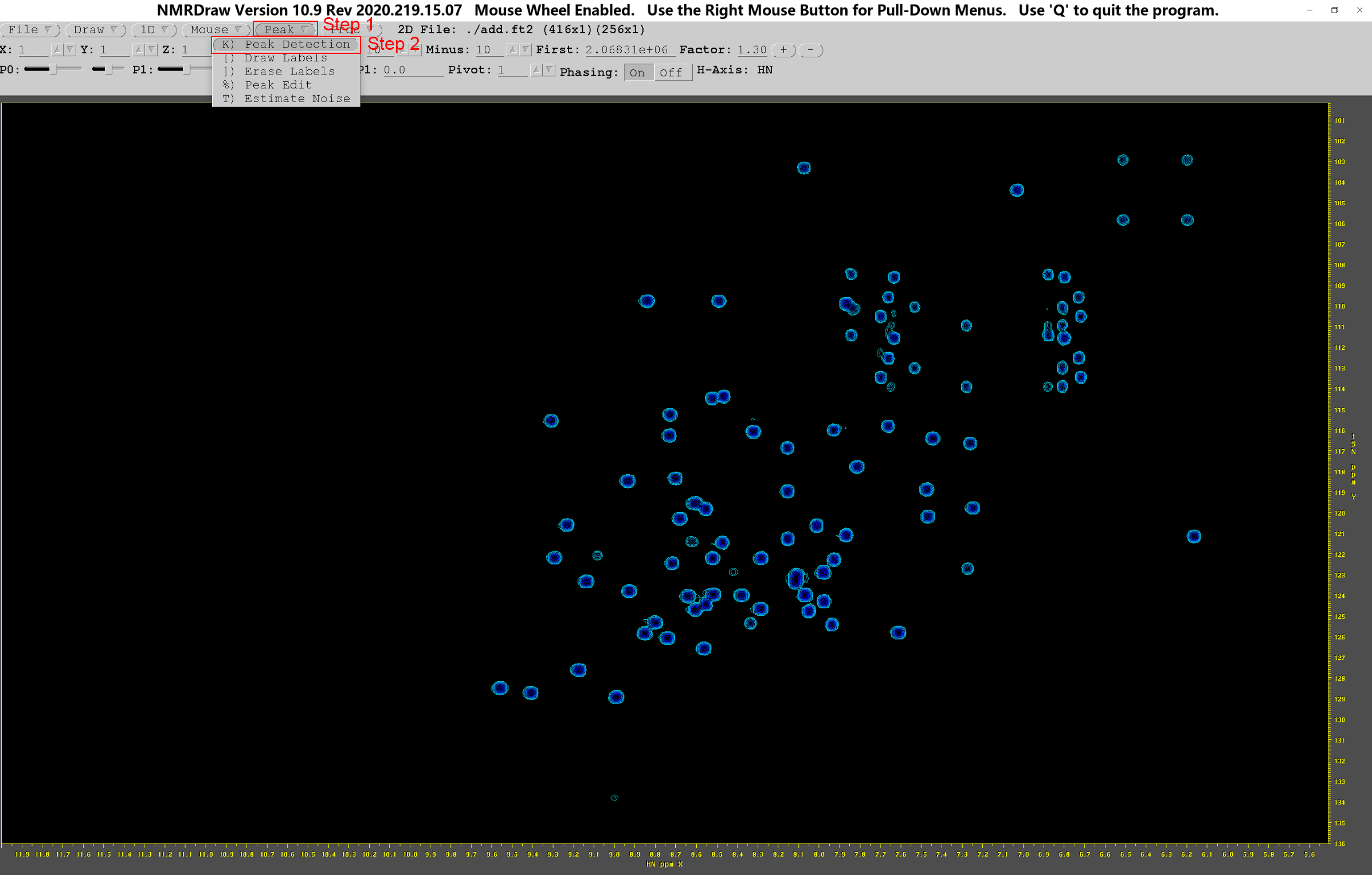
此时便会弹出“Peak Detection”对话框,然后单击“Detect”按钮,标出图中所有峰的序号,之后更改峰序号表为“test_add.tab”,点击“Save”保存后即可退出。
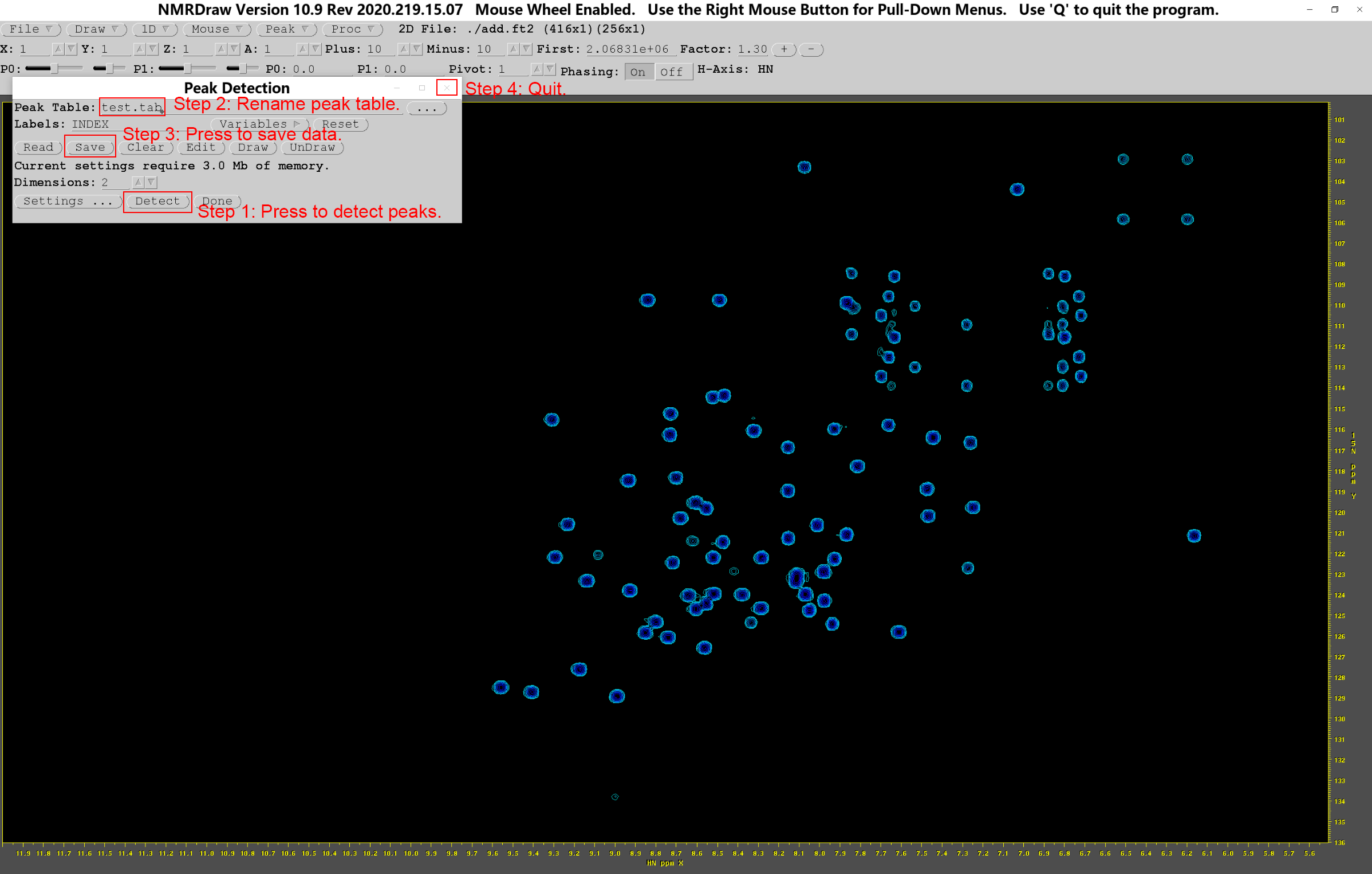
用同样的方法,可以得到sub.ft2的峰序号表“test_sub.tab”,之后就可以比照标准图谱,进行峰的指认。
5.峰的指认
为完成峰的指认,通常需要准备一张已经标好信号峰与其归属残基的标准图谱。此处以泛素蛋白为例,其标准图谱的归属保存于wtUb_ass.tab,放置在与add.ft2和sub.ft2所处的文件夹下。我们需要在终端输入命令:
ipap.tcl -inName1 test_add.tab -specName1 add.ft2 -assName wtUb_ass.tab -single
这样就可以开始对add.ft2的指认,此时会跳出如下窗口:
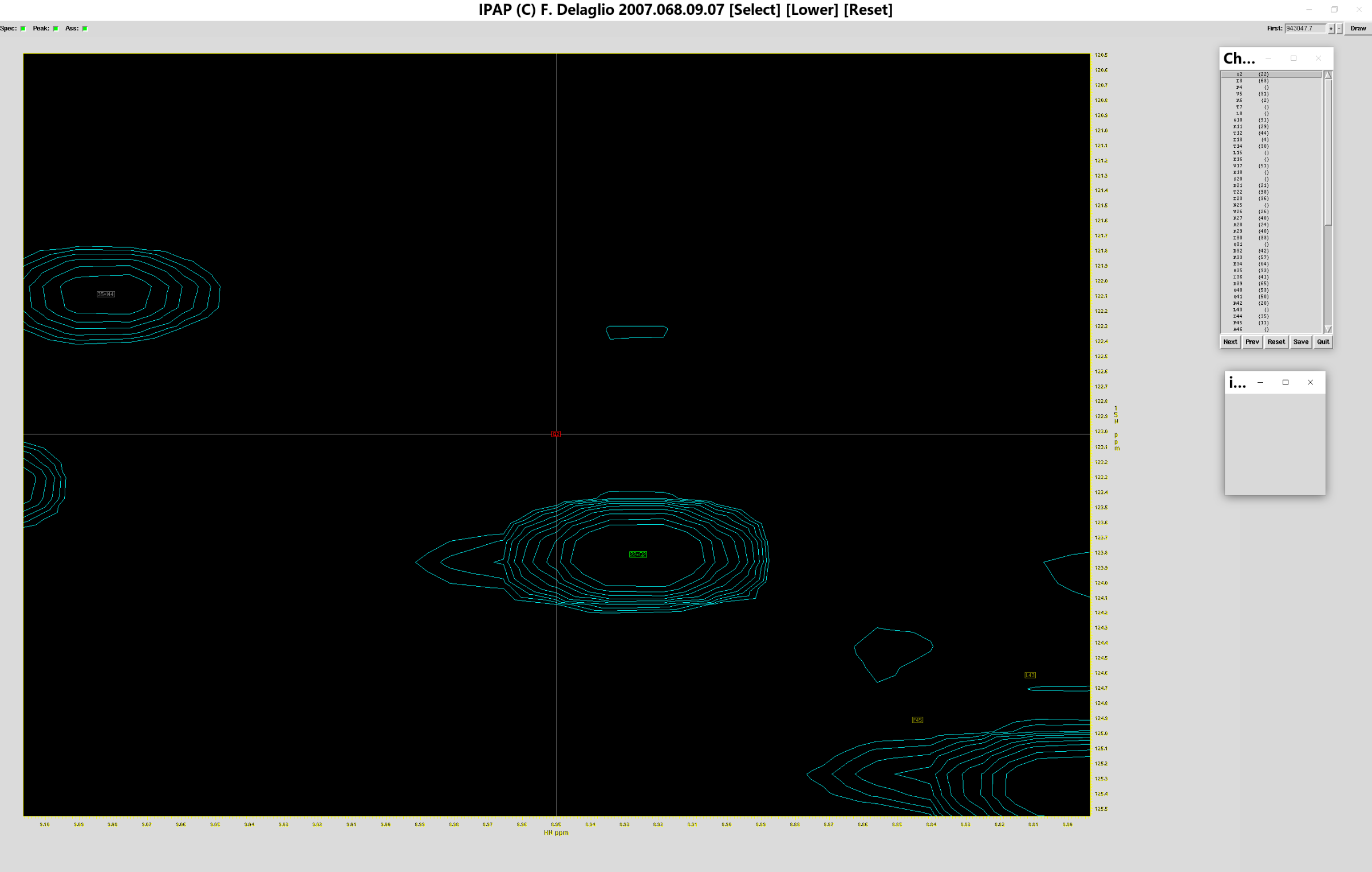
可以看到指认界面由若干部分组成:指认表(第一列为标准图谱中残基氨基酸代号和在多肽中的序号,第二列为测定图谱的序号,点击Next和Prev会跳转到上一个标准图谱序号对应的位置),指认图谱(含标准图谱和测定图谱的序号,红色方框为当前标准图谱序号对应的位置,绿色方框为测定图谱中指认的对应峰的位置)
由于指认图谱只显示一小部分,因此可以调节x轴和y轴的比例尺,使谱图显示得更为完整。例如输入以下命令并执行:
ipap.tcl -inName1 test_add.tab -specName1 add.ft2 -assName wtUb_ass.tab -single -xv 1 -yv 3
此时会跳出如下窗口:
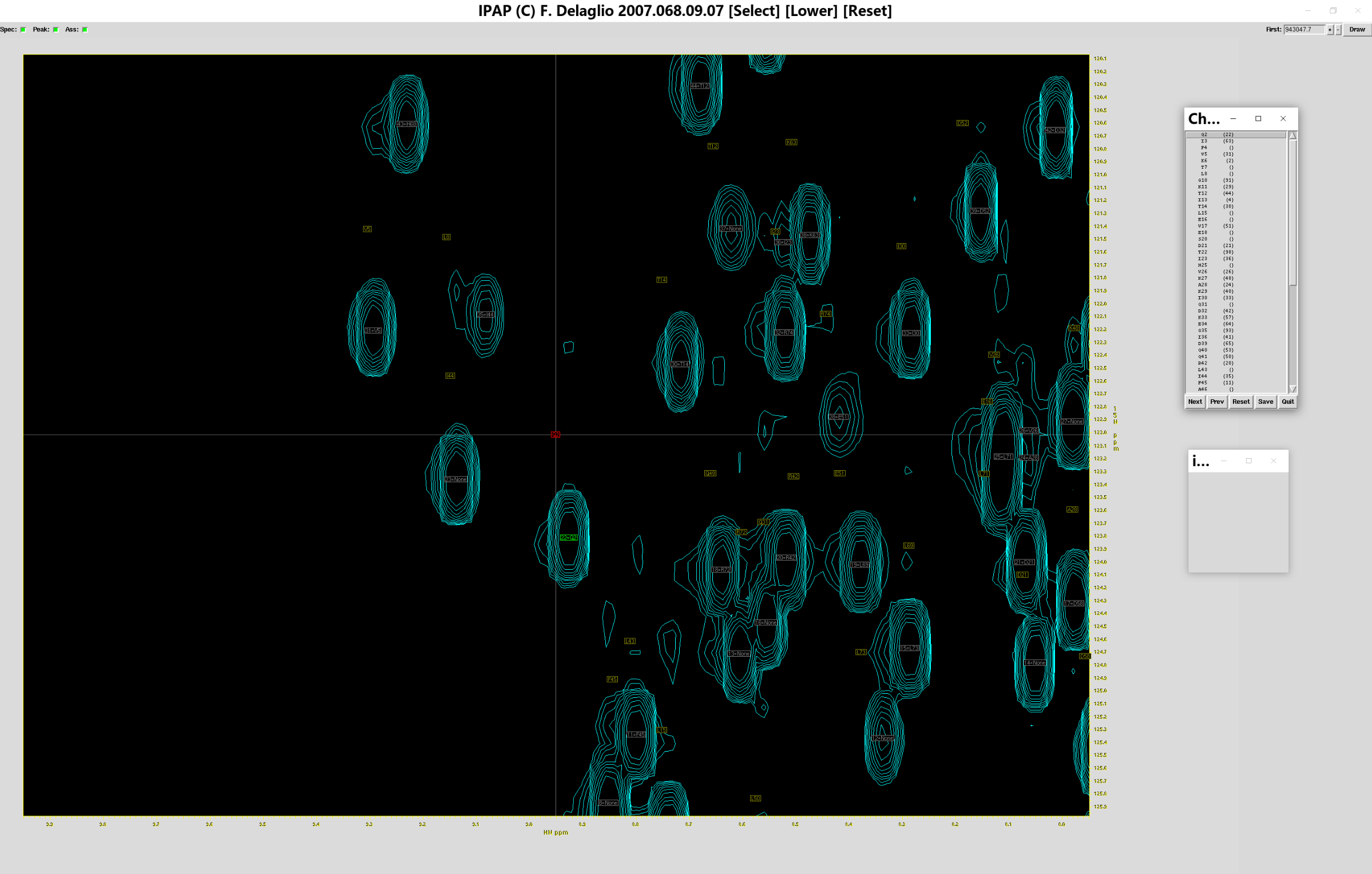
根据周围几个残基位移的大小,我们可以挪移标准图谱,使其与测定图谱对齐,例如输入以下命令并执行:
ipap.tcl -inName1 test_add.tab -specName1 add.ft2 -assName wtUb_ass.tab -single -xOff -0.025 -yOff 0.8
此时会跳出如下窗口:
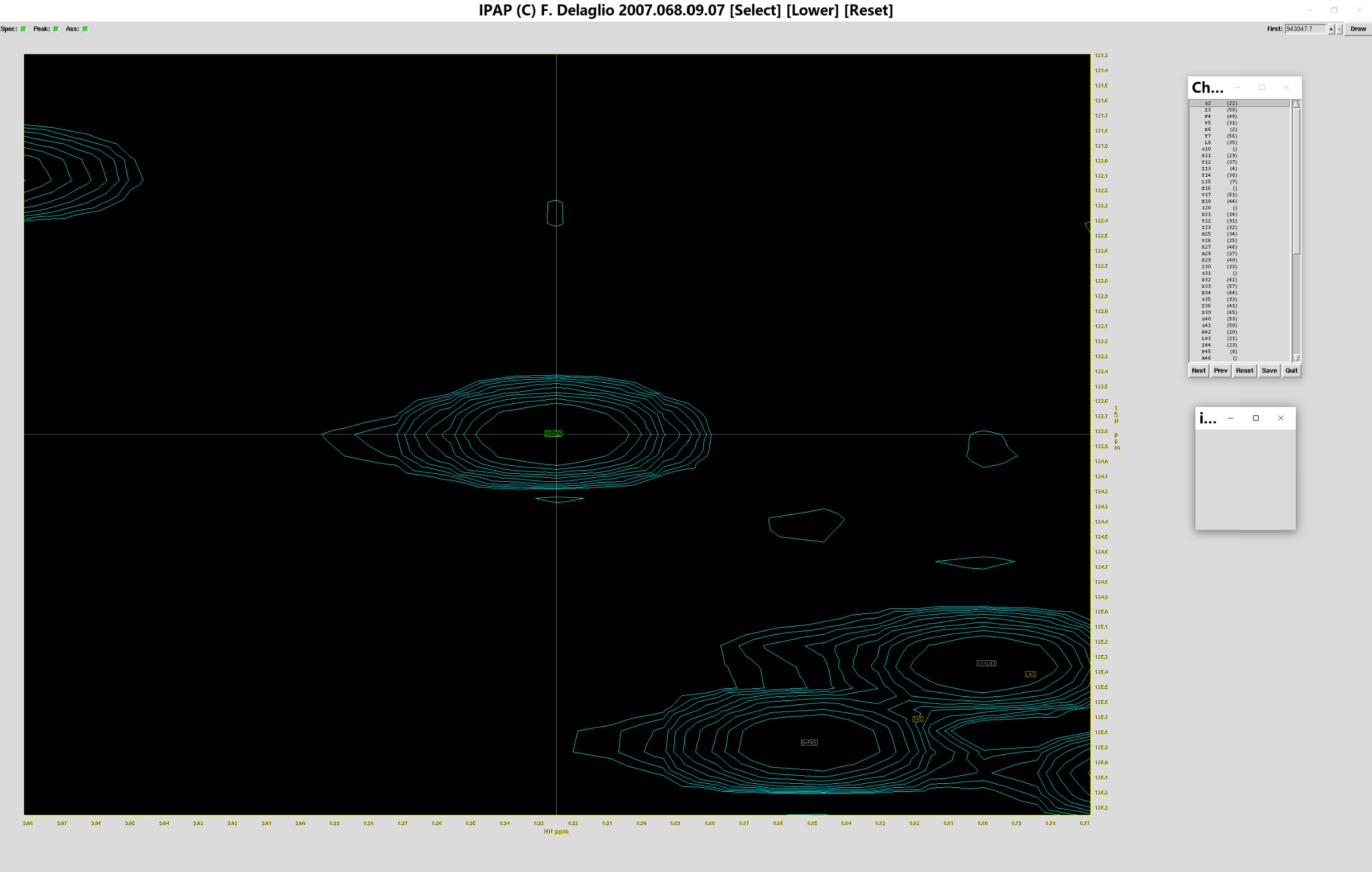
可以看到红色方框与绿色方框已经对准,此时点击指认表的“Next”,查看其余残基归属是否正确,如果某个残基缺乏归属,便不会出现对应的绿框,如下图所示
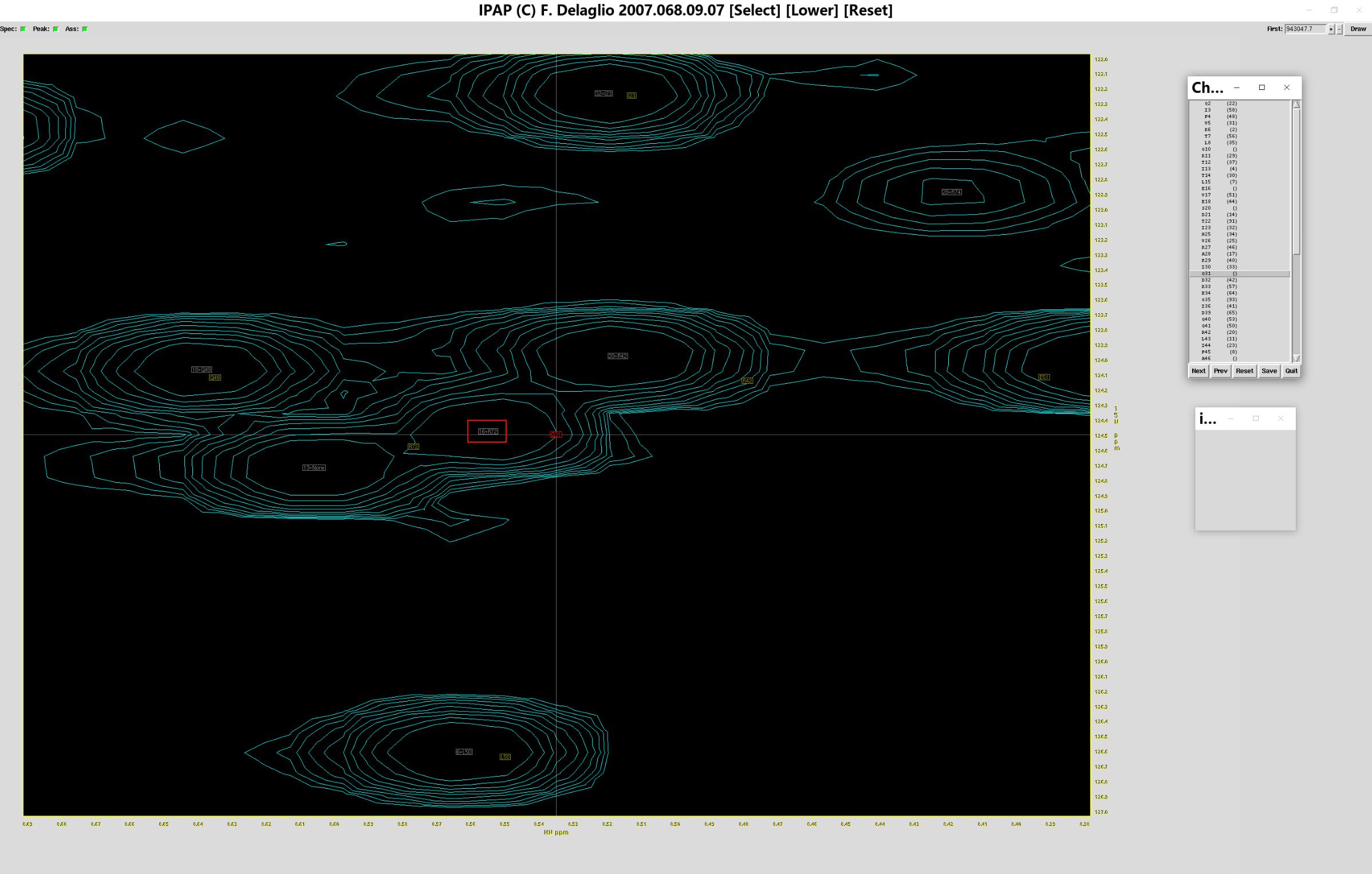
此时点击(你认为正确的归属位置对应的)灰色方框(如上图大红框所示),就可以将标准谱图的峰归属到测定谱图中“正确”的位置上,相应的,灰色方框就会变成绿色方框,代表红色小方框对应的残基标准位置,在实验谱图中对应于绿色小方框的位置
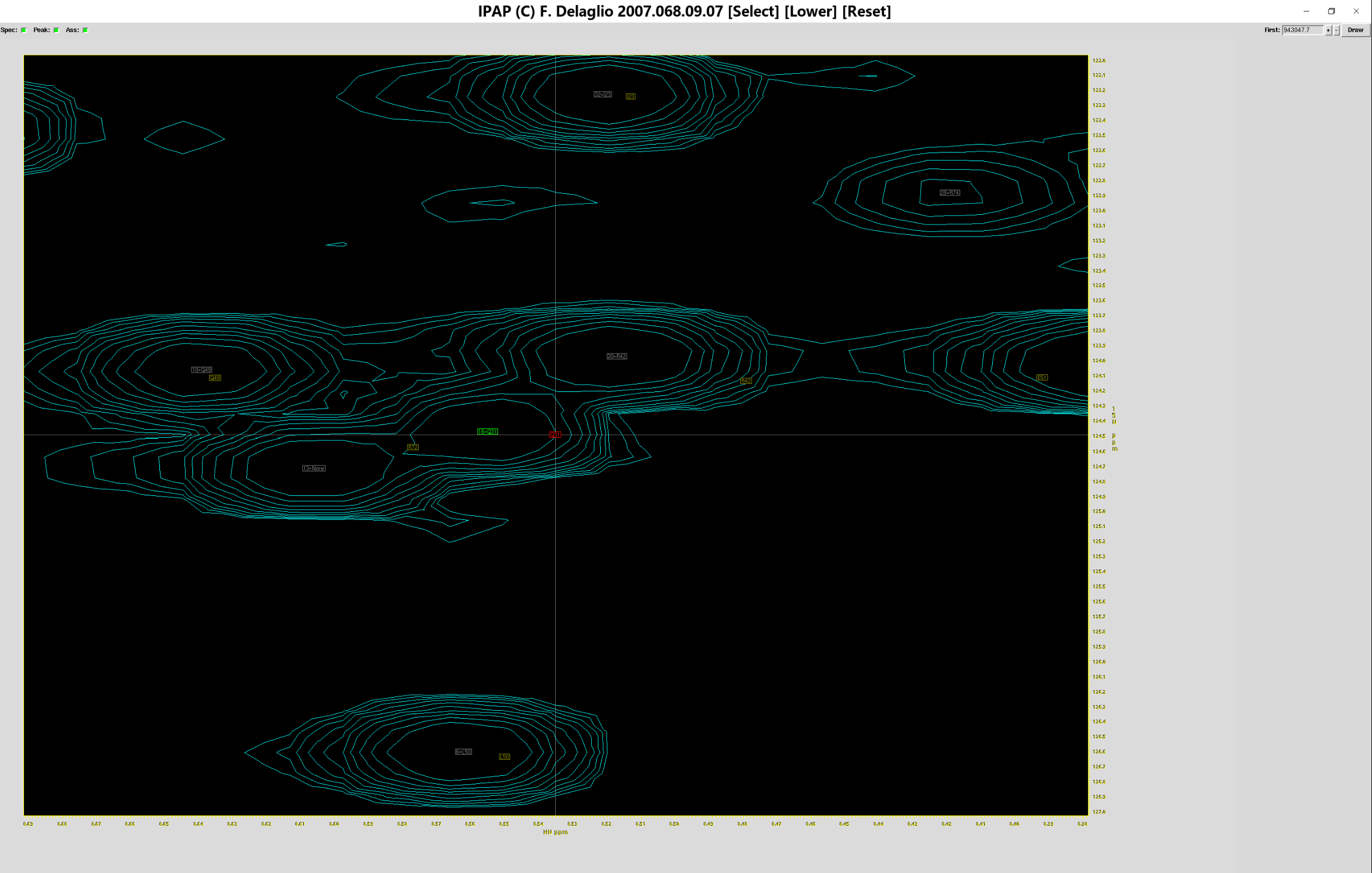
确认所有峰归属到正确位置上后(当然,有些标准图谱的峰在测定图谱中没有出现,这样的峰可以跳过),点击指认表的按钮“Save”,弹出保存窗口,重命名归属结果为sum.add_ass.tab,点击“Save”保存,之后即可退出指认窗口。用同样的方法,指认sub.ft2,将归属结果保存于sum.sub_ass.tab。
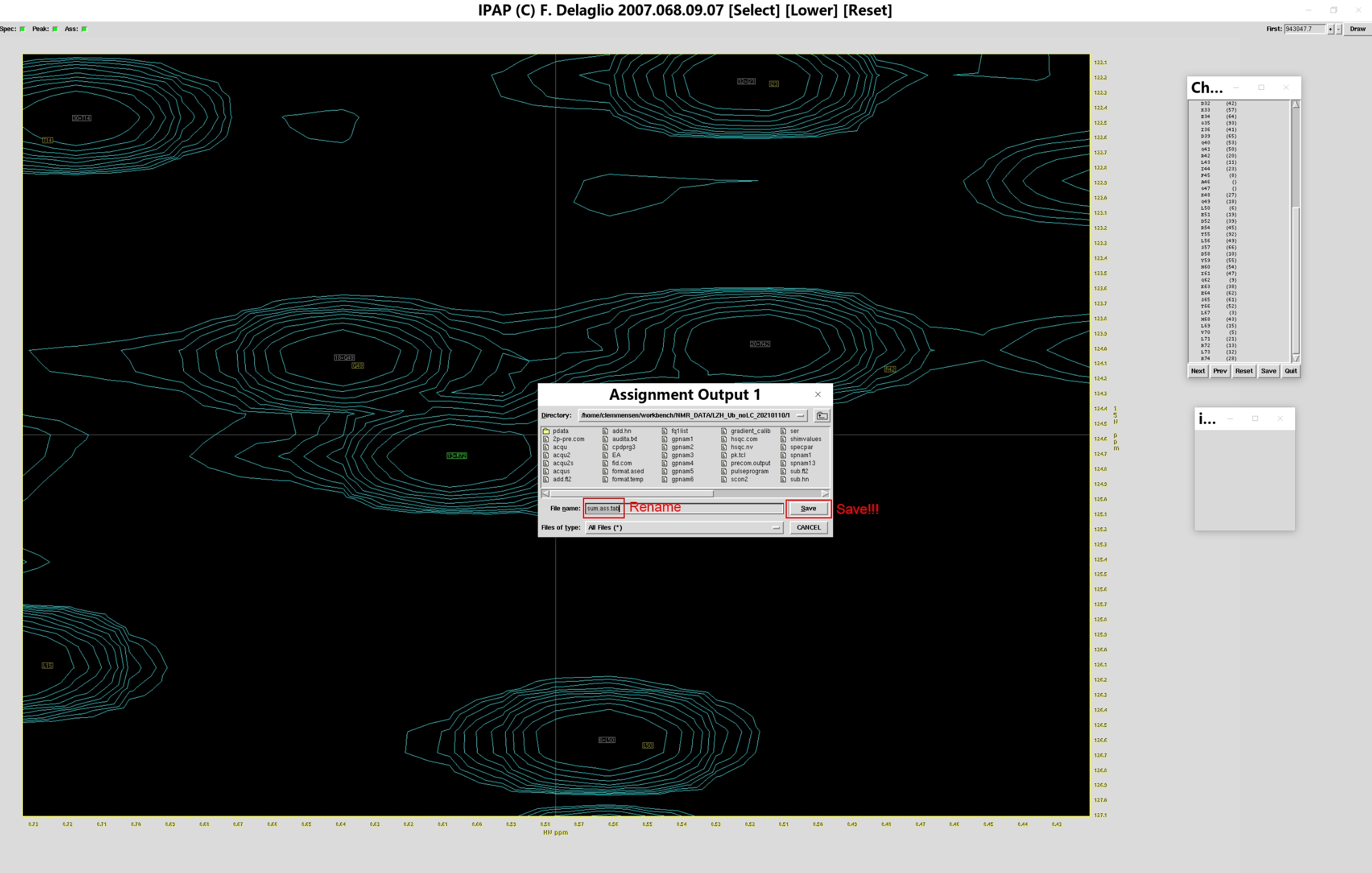
后记:之前听老师讲解ipap.tcl的使用,突然发现只需输入如下命令,便能实现在同一界面下,同时归属IPAP拆分的两张谱。此时,标准图谱的峰最好位于高场分量与低场分量的正中间,便于归属时对照方便。要注意的是,使用该命令打开ipap.tcl后,保存时会提示先保存第一张谱的归属文件,然后提示保存第二张谱的归属文件,对于下列命令,意味着先保存add.ft2对应的归属文件,后保存sub.ft2对应的归属文件。
ipap.tcl -inName1 test_add.tab -specName1 add.ft2 -inName2 test_sub.tab -specName2 sub.ft2 -assName wtUb_ass.tab -single -xOff -0.025 -yOff 0.8
此外,归属时有个小技巧:对于已经标记(显示为绿色方框)但是很明显对应错误的峰,可以用右键单击的方式将其取消,此时绿色方框会变成灰色方框,且原有的“等号+残基名”的标记将会消失,代表这个实验测出来的峰清空了原有归属。
6. 峰归属的输出
在当前终端输入以下命令并执行
grep -v one sum.add_ass.tab > temp2_add.tab
grep -v one sum.sub_ass.tab > temp2_sub.tab
awk '{if (NR>4) print $23,$9 }' temp2_add.tab | sort -nk1 > add.hn
awk '{if (NR>4) print $23,$9 }' temp2_sub.tab | sort -nk1 > sub.hn
得到简化的峰归属表add.hn和sub.hn,它记录了每个残基的-NH-中N的化学位移
7. RDC数据的整理
(处理前一定要按照第1-6部分,将各向同性溶液的IPAP和各向异性溶液的IPAP处理好,得到对应的峰归属数据!)
用Execl打开各向同性溶液的add.hn和sub.hn,将add.hn或sub.hn的第一列,以及add.hn和sub.hn各自的第二列,复制到新的工作簿中,然后两两相减,存于第四列,得1_control.xlsx;对各向异性溶液的add.hn和sub.hn也作相似处理,得2_exp.xlsx。
然后将2_exp.xlsx的第四列与1_control.xlsx的第四列相减,得新列,存于delta.xlsx,取出残基名称列和新列,并将残基名称列的字母删去,另存为delta.hn。
注意:add.hn和sub.hn一定要保证峰与峰数据对齐,否则可能会多一行或少一行数据,导致部分数据无法正确相减!
III. 用RDC数据验证解析结构与实际结构的一致性
将RDC格式转换脚本rdc_format.sh、RDC数据delta.hn、用于RDC理论效应计算的泛素蛋白结构文件1ubq_ambH.pdb拷贝至原始图谱的根目录中,输入以下命令并运行,以生成RDC张量分析所需的输入文件:
./rdc_format.sh delta.hn > rdc.tbl
再输入以下命令进行分析,并作出拟合曲线(需要预先安装Xplor-NIH):
calcTensor rdc.tbl 1ubq_ambH.pdb -plot
结果如下图所示:
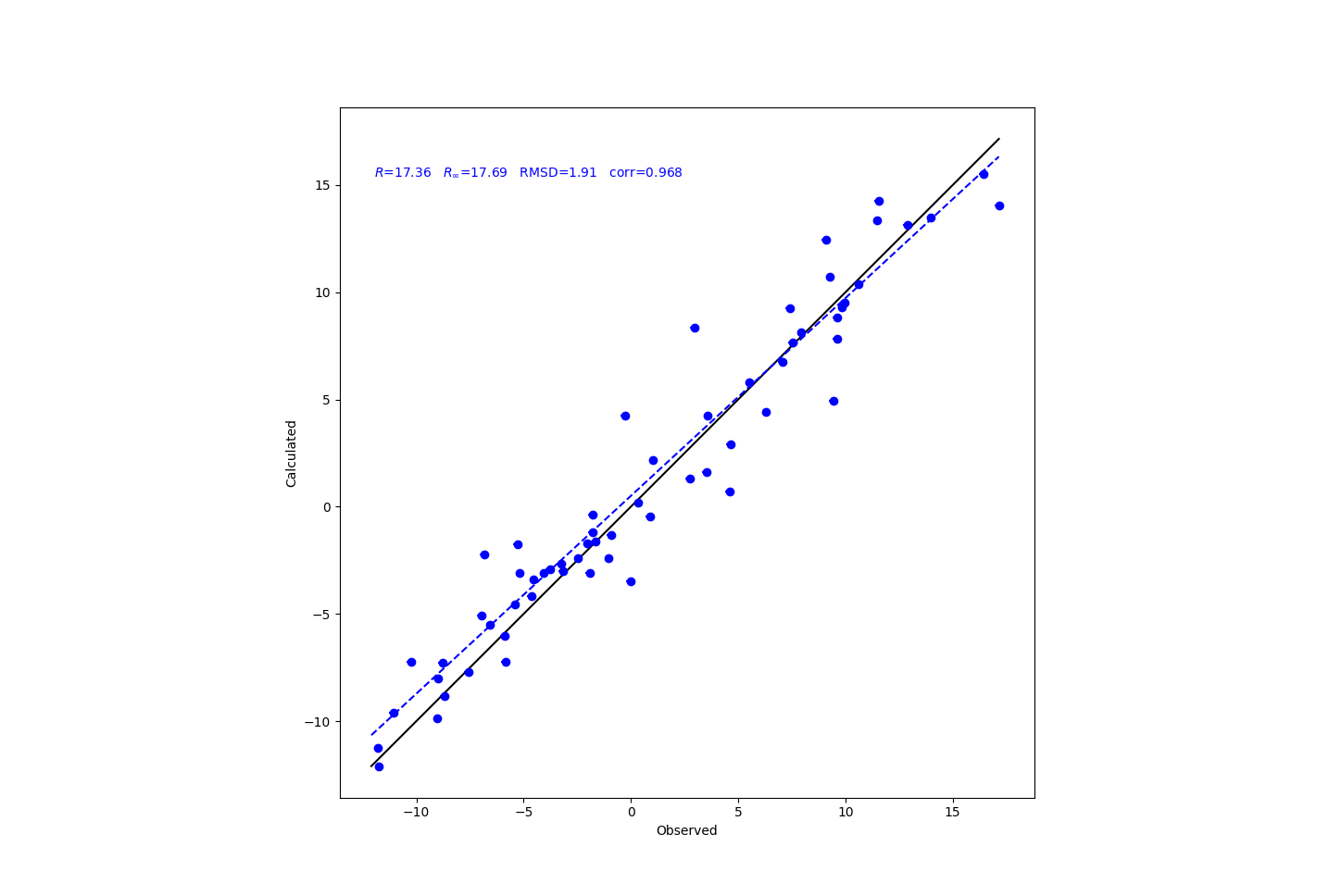
以下命令将给出具体数据,只要将终端输出的信息复制下来,即可用于后期拟合:
calcTensor rdc.tbl 1ubq_ambH.pdb -showRDCs
IV. 补充:Xplor-NIH的安装
直接解压缩法
1.安装包下载:访问Xplor-NIH并登录,若为初次访问,按照其提示注册账号(邮箱最好采用学校邮箱)。登陆后下载最新版本的Xplor-NIH,它包括xplor-nih-VERSION-Linux_x86_64.tar.gz(x86_64版Linux的Xplor-NIH)和xplor-nih-VERSION-db.tar.gz(数据库和示例),VERSION为当前版本号。
2.解压缩:创建一个文件夹(如/home/(你的用户名)/Xplor/opt),并将xplor-nih-VERSION-Linux_x86_64.tar.gz和xplor-nih-VERSION-db.tar.gz移入该文件夹中,打开终端,输入:
tar -xzvf xplor-nih-VERSION-Linux_x86_64.tar.gz
tar -xzvf xplor-nih-VERSION-db.tar.gz
这样就将Xplor-NIH解压到文件夹/home/(你的用户名)/Xplor/opt/xplor-nih-VERSION中
3.配置Xplor-NIH:输入以下命令
cd xplor-nih-VERSION
./configure
4.后续调整:以管理员(root)身份登录,在/home/(你的用户名)/Xplor/opt/xplor-nih-VERSION打开终端,输入./configure -symlinks DIR,回车。其中DIR为\$PATH所在位置,如\$HOME/bin或/usr/local/bin,可以用echo \$PATH获取。
5.测试Xplor运行情况:以普通用户身份登录,在/home/(你的用户名)/Xplor/opt/xplor-nih-VERSION打开终端,输入bin/testDist,回车。
脚本安装法
1.脚本下载:访问Xplor-NIH并登录,下载最新版本的installer-linux-VERSION.sh,并拷贝至/home/(你的用户名)/Xplor/opt
2.运行安装脚本:打开终端,输入chmod 777 installer-linux-VERSION.sh,回车(有GUI者,右键点属性→权限,全部修改为“允许读取、写入和执行”),然后输入./installer-linux-VERSION.sh,回车,等待压缩包全部下载并解压。
3.后续调整和测试:同“直接解压缩法”第4、5步。
This blog is under a CC BY-NC-SA 4.0 License
本文链接:http://chemlzh.github.io/2022/11/17/RDC-data-processing-tutorial/